
NHS England North West GMSA - IHE Laboratory Testing Workflow (LTW)
0.0.1 - ci-build
![]()
NHS England North West GMSA - IHE Laboratory Testing Workflow (LTW)
0.0.1 - ci-build
![]()
DRAFT Implementation Guide
This is for collaboration and discussion purposes and is subject to change.
NHS England North West GMSA - IHE Laboratory Testing Workflow (LTW) - Local Development build (v0.0.1) built by the FHIR (HL7® FHIR® Standard) Build Tools. See the Directory of published versions
| Official URL: https://fhir.north-west.england.nhs.uk/Questionnaire/81247-9 | Version: 0.0.1 | |||
| Draft as of 2025-04-20 | Computable Name: | |||
Master HL7 genetic variant reporting panel Questionnaire Viewer
Generated Narrative: Questionnaire 81247-9
Structure| LinkID | Text | Cardinality | Type | Description & Constraints |
|---|---|---|---|---|
| Questionnaire | https://fhir.north-west.england.nhs.uk/Questionnaire/81247-9#0.0.1 | |||
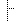 | Overall study variables type | 0..1 | group | |
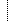 | Reason for study | 0..1 | string | |
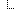 | The freeform text that is entered by the ordering provider to further annotate the coded Reason for Study [LOINC: 51967-8] associated with an ordered test. In HL7 v2 messages, OBR-31 should be used to report the reason for study. | 0..1 | display | |
| Gene dis assessed | 0..1 | string | |
| Coded identifier of the disorder being assessed but with exception to allow the recording of something not included in the controlled vocabulary that is being used. Various coding systems may be used, including ICD-9-CM, ICD-10-CM, SCT and NCBI MedGen. | 0..1 | display | |
| Medication assessed | 0..1 | string | |
| A coded medication assessed in a pharmacogenic test (recommend RxNorm) | 0..1 | display | |
| Gene studied ID | 0..1 | string | |
| HUGO Gene Nomenclature Committee (HGNC) identifier for a gene. List the gene(s) examined in full or in part by the study. If the study addresses multiple genes, these can be recorded in one OBX as a list seperated by repeat delimiters or in mulltiple OBX's with one gene per OBX. The recommended coding system will use the HGNC gene symbol as the display text and HGNC gene ID as the code. For example, 21497^ACAD9^HGNC. | 0..1 | display | |
| Gene Mut Tested Bld/T | 0..1 | string | |
| For targeted mutation analysis, report the discrete mutations the study is designed to detect. In HL7 V2 they can be reported in one observation as a list separated by repeat delimiters OR as a series of separate OBX segments, one per mutation tested for. In FHIR, multiple coded entries can not be reported as a list in one observation value field. They must be reported as the values of separate observations. | 0..1 | display | |
| DNA region of interest NumRange | 0..1 | string | |
| This term is used to report the region(s) of interest for sequencing studies as one or more numeric ranges that identify the parts of the reference sequence that are sequenced. These can be recorded as one or more HL7 numeric ranges using repeat delimiters to seperate multiple such ranges. They can also be recorded singly, one per OBX, using OBX-4 to distinguish these repeats with the same Observation ID. However, such detailed specification of the sequencing region of interest is rare, in part because this information is often proprietary, and the region of interest is reported as a text description instead, e.g., "Sequenced all of the coding, and appropriate flanking regions," using [LOINC: 81293-3]. | 0..1 | display | |
| DNA range(s) examined Nar | 0..1 | string | |
| This term is used to report a narrative description of the range(s) of DNA sequences examined in this sequencing study. Genetic test reports only rarely include explicit numeric ranges (which would be reported using [LOINC: 51959-5]) beause they are often proprietary, and more often describe the regions examined in narrative. For example, "all coding regions and appropriate flanking regions." To report the region of interest (e.g., in terms of introns and exons) rather than the specific DNA sequences examined, [LOINC: 47999-8] may be used. | 0..1 | display | |
| Gene dis anl interp-Imp | 0..1 | choice | Options: 4 options |
| Interpretation of all identified DNA Markers and/or Individual Alleles along with any known clinical information for the benefit of aiding clinicians in understanding the results overall. This is used for Symptomatic or Asymptomatic testing other than Carrier testing. | 0..1 | display | |
| Del-dup interp Patient-Imp | 0..1 | choice | Options: 3 options |
| Gene analysis narr rpt Doc | 0..1 | string | |
| Narative report in disease diagnostic-based format. | 0..1 | display | |
| Struct var ISCN name | 0..1 | string | |
| ISCN is a syntax for describing cytogenetic findings, from classical karyotypes to details that can be observed with copy number methodologies. Using ISCN nomenclature is highly recommended for reporting structural variants. | 0..1 | display | |
| Human ref seq assembly+build | 0..1 | choice | Options: 5 options |
| The NCBI build id for human genome assemblies. | 0..1 | display | |
| HGVS version | 0..1 | string | |
| Report the version of HGVS used for all observations specified using HGVS nomenclature. Any change in the HGVS recommendations will get a new version number based on the date of the change. The format for reporting the HGVS version used is: <version #>.<date produced in YYMMDD format>, for example, 2.120831. | 0..1 | display | |
| dbSNP version | 0..1 | string | |
| COSMIC version | 0..1 | string | |
| ClinVar version | 0..1 | string | |
| Simple var pnl | 0..1 | group | |
| Variant category | 0..1 | choice | Options: 2 options |
| Simple var ID | 0..1 | string | |
| This term is used to report the unique identifier of the simple variant found in this study. The identifier may come from various sources, including NCBI's ClinVar and Ensembl. For example, the variant NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys) has the ClinVar ID 30880 and would be reported in OBX-5 as 30880^NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys)^ClinVar. [http://www.ncbi.nlm.nih.gov/clinvar/variation/30880/] | 0..1 | display | |
| Gene studied ID | 0..1 | string | |
| HUGO Gene Nomenclature Committee (HGNC) identifier for a gene. List the gene(s) examined in full or in part by the study. If the study addresses multiple genes, these can be recorded in one OBX as a list seperated by repeat delimiters or in mulltiple OBX's with one gene per OBX. The recommended coding system will use the HGNC gene symbol as the display text and HGNC gene ID as the code. For example, 21497^ACAD9^HGNC. | 0..1 | display | |
| Transcript ref sequence ID | 0..1 | string | |
| This field carries the ID for the transcribed reference sequence, which is the part of the genomic reference sequence that is converted to messenger RNA (i.e., after the introns are removed). The transcript reference sequence ID may be reporting using various coding systems including NCBI's RefSeq ("NM_..."), Ensembl ("ENST..."), and LRG ("LRG..." plus "t1" to indicate transcript). | 0..1 | display | |
| DNA change | 0..1 | string | |
| Human Genome Variation Society (HGVS) nomenclature for a single DNA marker. The use of the nomenclature must be extended to describe non-variations (aka. wild types) see samples for wild type examples. | 0..1 | display | |
| Amino acid change | 0..1 | string | |
| Human Genome Variation Society (HGVS) nomenclature for an amino acid sequence. This value is derivable from the DNA Marker value if available. It is provided for convenience. The use of the nomenclature must be extended to describe non-variations (aka. wild types) see samples for wild type examples. | 0..1 | display | |
| DNA Change Type | 0..1 | choice | Options: 17 options |
| Codified type for associated DNA Marker. DNA Marker's use the HGVS notation which implies the DNA Marker Type, but the concurrent use of this code will allow a standard and explicit type for technical and display convenience. | 0..1 | display | |
| Amino acid change type | 0..1 | choice | Options: 11 options |
| Codified type for associated Amino Acid Marker. Amino Acid Marker's use the HGVS notation which implies the Amino Acid Marker Type, but the concurrent use of this code will allow a standard and explicit type for technical and display convenience. | 0..1 | display | |
| Genomic reference sequence ID | 0..1 | string | |
| This field carries the ID for the genomic reference sequence. The genomic reference sequence is a contiguous stretch of chromosome DNA that spans all of the exons of the gene and includes transcribed and non transcribed stretches. For this ID use either the NCBI genomic nucleotide RefSeq IDs with their version number (see: NCBI.NLM.NIH.Gov/RefSeq) or use the LRG identifiers, without transcript (t or p) extensions -- when they become available. (See- Report sponsored by GEN2PHEN at the European Bioinformatics Institute at Hinxton UK April 24-25, 2008). The NCI RefSeq genomic IDs are distinguished by a prefix of"NG" for genes from the nuclear chromosomes and prefix of "NC" for genes from mitochondria. The LRG Identifiers have a prefix of "LRG_" Mitochondrial genes are not in the scope of LRG | 0..1 | display | |
| Struct var HGVS name | 0..1 | string | |
| The name of a structural variant reported using HGVS nomenclature. | 0..1 | display | |
| Ref nucleotide | 0..1 | string | |
| Reference values ("normal") examined within the Reference Sequence. This is used in a genotyping test to define the reference and variable nucleotide strings. That is if the sequence variation is an insertion, then Reference Nucleotide will be blank and Variable Nucleotide will contain the inserted nucleotides. In contrast, if the sequence variation is a deletion, then the Reference Nucleotide will contain the deleted nucliotieds, and the Variable Nucleotide will be blank. | 0..1 | display | |
| Gen allele loc ID | 0..1 | string | |
| The variant start-end location is the first genomic position in the reference allele that contains a change from the reference allele. For example, for the simple variant NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys), the variant exact start-end location is Chr3: 128906220 on Assembly GRCh38. [http://www.ncbi.nlm.nih.gov/clinvar/variation/30880/] | 0..1 | display | |
| Alt allele | 0..1 | string | |
| The genomic alternate allele is the contiguous segment of DNA in the test sample that differs from the reference allele at the same location and thus defines a variant. | 0..1 | display | |
| Haplotype name Bld/T | 0..1 | string | |
| dbSNP ID | 0..1 | string | |
| The unique identifier for the variant represented as a small nucleotide polymorphism (SNP). The dbSNP ID is used routinely as the base identifier in pharmacogenomics as well as arrCGH studies. For example, for the simple variant NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys), the dbSNP ID is 368949613. [http://www.ncbi.nlm.nih.gov/clinvar/variation/30880/] | 0..1 | display | |
| CIGAR var ID | 0..1 | string | |
| This term is used to report the unique ID from CIGAR, a syntax for describing variation that is use most frequently during alignment in sequencing studies. | 0..1 | display | |
| Cyto loc ID | 0..1 | string | |
| Genomic source class | 0..1 | choice | Options: 8 options |
| The genomic class of the specimen being analyzed: Germline for inherited genome, somatic for cancer genome, and prenatal for fetal genome. | 0..1 | display | |
| Struct var analysis method | 0..1 | choice | Options: 23 options |
| The method used for analyzing chromosome structural variation, such as FISH, arrCGH, sequencing, and MLPA. | 0..1 | display | |
| Gene dis seq var interp-Imp | 0..1 | choice | Options: 5 options |
| Single DNA marker or individual allele interpretation in the context of the assessed genetic disease. Prior to the LOINC release 2.56 (June 2016), the answer list was updated per the recommendations of the American College of Medical Genetics (ACMG). The previous answer list number was LL603-2, and two of the answer strings and LA codes are the same in the new list (pathogenic and benign). In the new answer list, the presumed pathogenic, unknown significance and presumed benign answers from LL603-2 have been replaced by likely pathogenic, uncertain significance and likely benign. The answer strings and their respective LA codes from LL603-2 remain valid. | 0..1 | display | |
| Genetic var assess | 0..1 | choice | Options: 4 options |
| Where testing scenarios are intended to assess the prescence or absence of a known set of DNA variants (e.g. tumor profiling using genotyping technology), then the Genetic Variant Assessment is used in conjunction with answer list supports structured communication of these findings. Of note, 'No Call' is different from 'Absent', because 'No Call' did not result in the determination of the marker's presents or absents. This may be due to test failure or specimen specific context which renders the test ineffective. | 0..1 | display | |
| Prob assoc phenotype | 0..1 | string | |
| The possible phenotype associated with the genetic variant found in this study. | 0..1 | display | |
| Allelic state | 0..1 | choice | Options: 5 options |
| The level of occurrence of a single DNA Marker within a set of chromosomes. Heterozygous indicates the DNA Marker is only present in one of the two genes contained in homologous chromosomes. Homozygous indicates the DNA Marker is present in both genes contained in homologous chromosomes. Hemizygous indicates the DNA Marker exists in the only single copy of a gene in a non-homologous chromosome (The male X and Y chromosome are non-homologous). Hemiplasmic indicates that the DNA Marker is present in some but not all of the copies of mitochondrial DNA. Homoplasmic indicates that the DNA Maker is present in all of the copies of mitochondrial DNA. | 0..1 | display | |
| Sample VAF | 0..1 | decimal | |
| The fraction of all reads in a study sample at a given genomic locus that identify the allele (variant) in question. For homozygotes it will be close to 1.0; for heterozygotes it will be close to 0.5. It can be less than 0.5 in the case of mosaics or multiple chromosome, or mixtures of tumor cells and normal cells. This measure is an attribute of the variant and applies when the method is a Next Generation Sequencing (NGS) or similar. Such methods provide many reads from the sample for each locus. To report population allelic frequency, see [LOINC: 92821-8]. Allelic frequency is usually reported as a decimal fraction for both Sample Variant Allelic Frequency and Population Allelic Frequency, although it is occasionally reported as a percent. Special care/caution should be taken when reporting and converting to a decimal fraction. | 0..1 | display | |
| Allelic read depth | 0..1 | decimal | |
| Allelic phase | 0..1 | choice | Options: 9 options |
| Basis allelic phase | 0..1 | choice | Options: 4 options |
| This panel is used to report the information associated with a simple genetic variant, such as a single nucleotide change. It should not be used to report information related to structural variants. | 0..1 | display | |
| Struct variant pnl | 0..1 | group | |
| Gen struct var copy num | 0..1 | decimal | |
| Struct var rep arrCGH Rto | 0..1 | decimal | |
| Struct var len | 0..1 | decimal | |
| Length of the structural variant, which information may be ascertained in some but not all types of structural variants. | 0..1 | display | |
| Struct var outer start-end NumRange | 0..1 | decimal | |
| The genomic coordinates of the widest genomic range in which the variant might reside. | 0..1 | display | |
| Struct var inner start-end NumRange | 0..1 | decimal | |
| The genomic coordinates of the narrowest genomic range in which the variant might reside. | 0..1 | display | |
| Comp var pnl | 0..1 | group | |
| Comp var ID | 0..1 | string | |
| This term is used to report the unique identifier of the complex variant found in this study. The identifier may come from various sources, including NCBI's ClinVar and Ensembl. For example, the variant NM_000106.5(CYP2D6):c.[886C>T;457G>C] - Haplotype has the ClinVar ID 16895. [http://www.ncbi.nlm.nih.gov/clinvar/variation/16895/] | 0..1 | display | |
| Comp var HGVS name | 0..1 | string | |
| This term is used to report the name of the complex variant found in this study in HGVS format. For example, c.[886C>T;457G>C], which represents two separate base substitutions in one gene on one chromosome, or c.[886C>T];[457G>C], which represents two separate base substitutions in one gene on two different chromosomes. | 0..1 | display | |
| Comp var type | 0..1 | choice | Options: 4 options |
| The type of complex variant, for example, compound heterozygous or haplotype. | 0..1 | display | |
| Prob assoc phenotype | 0..1 | string | |
| The possible phenotype associated with the genetic variant found in this study. | 0..1 | display | |
| Gene dis seq var interp-Imp | 0..1 | choice | Options: 5 options |
| Single DNA marker or individual allele interpretation in the context of the assessed genetic disease. Prior to the LOINC release 2.56 (June 2016), the answer list was updated per the recommendations of the American College of Medical Genetics (ACMG). The previous answer list number was LL603-2, and two of the answer strings and LA codes are the same in the new list (pathogenic and benign). In the new answer list, the presumed pathogenic, unknown significance and presumed benign answers from LL603-2 have been replaced by likely pathogenic, uncertain significance and likely benign. The answer strings and their respective LA codes from LL603-2 remain valid. | 0..1 | display | |
| Allelic state | 0..1 | choice | Options: 5 options |
| The level of occurrence of a single DNA Marker within a set of chromosomes. Heterozygous indicates the DNA Marker is only present in one of the two genes contained in homologous chromosomes. Homozygous indicates the DNA Marker is present in both genes contained in homologous chromosomes. Hemizygous indicates the DNA Marker exists in the only single copy of a gene in a non-homologous chromosome (The male X and Y chromosome are non-homologous). Hemiplasmic indicates that the DNA Marker is present in some but not all of the copies of mitochondrial DNA. Homoplasmic indicates that the DNA Maker is present in all of the copies of mitochondrial DNA. | 0..1 | display | |
| Basis allelic phase | 0..1 | choice | Options: 4 options |
| Simple var pnl | 0..1 | group | |
| Variant category | 0..1 | choice | Options: 2 options |
| Simple var ID | 0..1 | string | |
| This term is used to report the unique identifier of the simple variant found in this study. The identifier may come from various sources, including NCBI's ClinVar and Ensembl. For example, the variant NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys) has the ClinVar ID 30880 and would be reported in OBX-5 as 30880^NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys)^ClinVar. [http://www.ncbi.nlm.nih.gov/clinvar/variation/30880/] | 0..1 | display | |
| Gene studied ID | 0..1 | string | |
| HUGO Gene Nomenclature Committee (HGNC) identifier for a gene. List the gene(s) examined in full or in part by the study. If the study addresses multiple genes, these can be recorded in one OBX as a list seperated by repeat delimiters or in mulltiple OBX's with one gene per OBX. The recommended coding system will use the HGNC gene symbol as the display text and HGNC gene ID as the code. For example, 21497^ACAD9^HGNC. | 0..1 | display | |
| Transcript ref sequence ID | 0..1 | string | |
| This field carries the ID for the transcribed reference sequence, which is the part of the genomic reference sequence that is converted to messenger RNA (i.e., after the introns are removed). The transcript reference sequence ID may be reporting using various coding systems including NCBI's RefSeq ("NM_..."), Ensembl ("ENST..."), and LRG ("LRG..." plus "t1" to indicate transcript). | 0..1 | display | |
| DNA change | 0..1 | string | |
| Human Genome Variation Society (HGVS) nomenclature for a single DNA marker. The use of the nomenclature must be extended to describe non-variations (aka. wild types) see samples for wild type examples. | 0..1 | display | |
| Amino acid change | 0..1 | string | |
| Human Genome Variation Society (HGVS) nomenclature for an amino acid sequence. This value is derivable from the DNA Marker value if available. It is provided for convenience. The use of the nomenclature must be extended to describe non-variations (aka. wild types) see samples for wild type examples. | 0..1 | display | |
| DNA Change Type | 0..1 | choice | Options: 17 options |
| Codified type for associated DNA Marker. DNA Marker's use the HGVS notation which implies the DNA Marker Type, but the concurrent use of this code will allow a standard and explicit type for technical and display convenience. | 0..1 | display | |
| Amino acid change type | 0..1 | choice | Options: 11 options |
| Codified type for associated Amino Acid Marker. Amino Acid Marker's use the HGVS notation which implies the Amino Acid Marker Type, but the concurrent use of this code will allow a standard and explicit type for technical and display convenience. | 0..1 | display | |
| Genomic reference sequence ID | 0..1 | string | |
| This field carries the ID for the genomic reference sequence. The genomic reference sequence is a contiguous stretch of chromosome DNA that spans all of the exons of the gene and includes transcribed and non transcribed stretches. For this ID use either the NCBI genomic nucleotide RefSeq IDs with their version number (see: NCBI.NLM.NIH.Gov/RefSeq) or use the LRG identifiers, without transcript (t or p) extensions -- when they become available. (See- Report sponsored by GEN2PHEN at the European Bioinformatics Institute at Hinxton UK April 24-25, 2008). The NCI RefSeq genomic IDs are distinguished by a prefix of"NG" for genes from the nuclear chromosomes and prefix of "NC" for genes from mitochondria. The LRG Identifiers have a prefix of "LRG_" Mitochondrial genes are not in the scope of LRG | 0..1 | display | |
| Struct var HGVS name | 0..1 | string | |
| The name of a structural variant reported using HGVS nomenclature. | 0..1 | display | |
| Ref nucleotide | 0..1 | string | |
| Reference values ("normal") examined within the Reference Sequence. This is used in a genotyping test to define the reference and variable nucleotide strings. That is if the sequence variation is an insertion, then Reference Nucleotide will be blank and Variable Nucleotide will contain the inserted nucleotides. In contrast, if the sequence variation is a deletion, then the Reference Nucleotide will contain the deleted nucliotieds, and the Variable Nucleotide will be blank. | 0..1 | display | |
| Gen allele loc ID | 0..1 | string | |
| The variant start-end location is the first genomic position in the reference allele that contains a change from the reference allele. For example, for the simple variant NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys), the variant exact start-end location is Chr3: 128906220 on Assembly GRCh38. [http://www.ncbi.nlm.nih.gov/clinvar/variation/30880/] | 0..1 | display | |
| Alt allele | 0..1 | string | |
| The genomic alternate allele is the contiguous segment of DNA in the test sample that differs from the reference allele at the same location and thus defines a variant. | 0..1 | display | |
| Haplotype name Bld/T | 0..1 | string | |
| dbSNP ID | 0..1 | string | |
| The unique identifier for the variant represented as a small nucleotide polymorphism (SNP). The dbSNP ID is used routinely as the base identifier in pharmacogenomics as well as arrCGH studies. For example, for the simple variant NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys), the dbSNP ID is 368949613. [http://www.ncbi.nlm.nih.gov/clinvar/variation/30880/] | 0..1 | display | |
| CIGAR var ID | 0..1 | string | |
| This term is used to report the unique ID from CIGAR, a syntax for describing variation that is use most frequently during alignment in sequencing studies. | 0..1 | display | |
| Cyto loc ID | 0..1 | string | |
| Genomic source class | 0..1 | choice | Options: 8 options |
| The genomic class of the specimen being analyzed: Germline for inherited genome, somatic for cancer genome, and prenatal for fetal genome. | 0..1 | display | |
| Struct var analysis method | 0..1 | choice | Options: 23 options |
| The method used for analyzing chromosome structural variation, such as FISH, arrCGH, sequencing, and MLPA. | 0..1 | display | |
| Gene dis seq var interp-Imp | 0..1 | choice | Options: 5 options |
| Single DNA marker or individual allele interpretation in the context of the assessed genetic disease. Prior to the LOINC release 2.56 (June 2016), the answer list was updated per the recommendations of the American College of Medical Genetics (ACMG). The previous answer list number was LL603-2, and two of the answer strings and LA codes are the same in the new list (pathogenic and benign). In the new answer list, the presumed pathogenic, unknown significance and presumed benign answers from LL603-2 have been replaced by likely pathogenic, uncertain significance and likely benign. The answer strings and their respective LA codes from LL603-2 remain valid. | 0..1 | display | |
| Genetic var assess | 0..1 | choice | Options: 4 options |
| Where testing scenarios are intended to assess the prescence or absence of a known set of DNA variants (e.g. tumor profiling using genotyping technology), then the Genetic Variant Assessment is used in conjunction with answer list supports structured communication of these findings. Of note, 'No Call' is different from 'Absent', because 'No Call' did not result in the determination of the marker's presents or absents. This may be due to test failure or specimen specific context which renders the test ineffective. | 0..1 | display | |
| Prob assoc phenotype | 0..1 | string | |
| The possible phenotype associated with the genetic variant found in this study. | 0..1 | display | |
| Allelic state | 0..1 | choice | Options: 5 options |
| The level of occurrence of a single DNA Marker within a set of chromosomes. Heterozygous indicates the DNA Marker is only present in one of the two genes contained in homologous chromosomes. Homozygous indicates the DNA Marker is present in both genes contained in homologous chromosomes. Hemizygous indicates the DNA Marker exists in the only single copy of a gene in a non-homologous chromosome (The male X and Y chromosome are non-homologous). Hemiplasmic indicates that the DNA Marker is present in some but not all of the copies of mitochondrial DNA. Homoplasmic indicates that the DNA Maker is present in all of the copies of mitochondrial DNA. | 0..1 | display | |
| Sample VAF | 0..1 | decimal | |
| The fraction of all reads in a study sample at a given genomic locus that identify the allele (variant) in question. For homozygotes it will be close to 1.0; for heterozygotes it will be close to 0.5. It can be less than 0.5 in the case of mosaics or multiple chromosome, or mixtures of tumor cells and normal cells. This measure is an attribute of the variant and applies when the method is a Next Generation Sequencing (NGS) or similar. Such methods provide many reads from the sample for each locus. To report population allelic frequency, see [LOINC: 92821-8]. Allelic frequency is usually reported as a decimal fraction for both Sample Variant Allelic Frequency and Population Allelic Frequency, although it is occasionally reported as a percent. Special care/caution should be taken when reporting and converting to a decimal fraction. | 0..1 | display | |
| Allelic read depth | 0..1 | decimal | |
| Allelic phase | 0..1 | choice | Options: 9 options |
| Basis allelic phase | 0..1 | choice | Options: 4 options |
| This panel is used to report the information associated with a simple genetic variant, such as a single nucleotide change. It should not be used to report information related to structural variants. | 0..1 | display | |
| This panel is used to report information related to a complex genetic variant and includes a repeating subpanel for reporting specific information for each simple variation that the complex variant includes. | 0..1 | display | |
| Pharmg result pnl | 0..1 | group | |
| Gene studied ID | 0..1 | string | |
| HUGO Gene Nomenclature Committee (HGNC) identifier for a gene. List the gene(s) examined in full or in part by the study. If the study addresses multiple genes, these can be recorded in one OBX as a list seperated by repeat delimiters or in mulltiple OBX's with one gene per OBX. The recommended coding system will use the HGNC gene symbol as the display text and HGNC gene ID as the code. For example, 21497^ACAD9^HGNC. | 0..1 | display | |
| Genotype name Patient | 0..1 | string | |
| Drug metab seq var interp-Imp | 0..1 | choice | Options: 5 options |
| Predicted phenotype for drug efficacy. A single marker interpretation value known to allow (responsive) or prevent (resistant) the drug to perform. Prior to the LOINC release 2.56 (June 2016), the answer list was updated per the recommendations of the Clinical Pharmacogenetics Implementation Consortium (CPIC). The previous answer list number was LL609-9, and three of the answer strings and LA codes are the same in the new list (ultrarapid metabolizer, intermediate metabolizer, and poor metabolizer). In the new answer list, the extensive metabolizer answer (LA10316-0) from LL609-9 has been replaced with two new answers (rapid metabolizer, normal metabolizer). LA10316-0 is still a valid LA code for the "extensive metabolizer" answer string. | 0..1 | display | |
| Drug eff seq var interp-Imp | 0..1 | choice | Options: 8 options |
| Predicted phenotype for ability of drug to bind to intended site in order to deliver intended affect. A single marker interpretation value known to allow (responsive) or prevent (resistant) the drug to perform. | 0..1 | display | |
| Genetic var eff high-risk allele | 0..1 | choice | Options: 2 options |
| Med usage impl pnl | 0..1 | group | |
| Medication assessed | 0..1 | string | |
| A coded medication assessed in a pharmacogenic test (recommend RxNorm) | 0..1 | display | |
| Med usage sugg | 0..1 | choice | Options: 5 options |
| Med usage sugg Patient-Imp | 0..1 | string | |
| Haplotype definition Pnl | 0..1 | group | |
| Gene studied ID | 0..1 | string | |
| HUGO Gene Nomenclature Committee (HGNC) identifier for a gene. List the gene(s) examined in full or in part by the study. If the study addresses multiple genes, these can be recorded in one OBX as a list seperated by repeat delimiters or in mulltiple OBX's with one gene per OBX. The recommended coding system will use the HGNC gene symbol as the display text and HGNC gene ID as the code. For example, 21497^ACAD9^HGNC. | 0..1 | display | |
| Haplotype name Bld/T | 0..1 | string | |
| Simple var pnl | 0..1 | group | |
| Variant category | 0..1 | choice | Options: 2 options |
| Simple var ID | 0..1 | string | |
| This term is used to report the unique identifier of the simple variant found in this study. The identifier may come from various sources, including NCBI's ClinVar and Ensembl. For example, the variant NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys) has the ClinVar ID 30880 and would be reported in OBX-5 as 30880^NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys)^ClinVar. [http://www.ncbi.nlm.nih.gov/clinvar/variation/30880/] | 0..1 | display | |
| Gene studied ID | 0..1 | string | |
| HUGO Gene Nomenclature Committee (HGNC) identifier for a gene. List the gene(s) examined in full or in part by the study. If the study addresses multiple genes, these can be recorded in one OBX as a list seperated by repeat delimiters or in mulltiple OBX's with one gene per OBX. The recommended coding system will use the HGNC gene symbol as the display text and HGNC gene ID as the code. For example, 21497^ACAD9^HGNC. | 0..1 | display | |
| Transcript ref sequence ID | 0..1 | string | |
| This field carries the ID for the transcribed reference sequence, which is the part of the genomic reference sequence that is converted to messenger RNA (i.e., after the introns are removed). The transcript reference sequence ID may be reporting using various coding systems including NCBI's RefSeq ("NM_..."), Ensembl ("ENST..."), and LRG ("LRG..." plus "t1" to indicate transcript). | 0..1 | display | |
| DNA change | 0..1 | string | |
| Human Genome Variation Society (HGVS) nomenclature for a single DNA marker. The use of the nomenclature must be extended to describe non-variations (aka. wild types) see samples for wild type examples. | 0..1 | display | |
| Amino acid change | 0..1 | string | |
| Human Genome Variation Society (HGVS) nomenclature for an amino acid sequence. This value is derivable from the DNA Marker value if available. It is provided for convenience. The use of the nomenclature must be extended to describe non-variations (aka. wild types) see samples for wild type examples. | 0..1 | display | |
| DNA Change Type | 0..1 | choice | Options: 17 options |
| Codified type for associated DNA Marker. DNA Marker's use the HGVS notation which implies the DNA Marker Type, but the concurrent use of this code will allow a standard and explicit type for technical and display convenience. | 0..1 | display | |
| Amino acid change type | 0..1 | choice | Options: 11 options |
| Codified type for associated Amino Acid Marker. Amino Acid Marker's use the HGVS notation which implies the Amino Acid Marker Type, but the concurrent use of this code will allow a standard and explicit type for technical and display convenience. | 0..1 | display | |
| Genomic reference sequence ID | 0..1 | string | |
| This field carries the ID for the genomic reference sequence. The genomic reference sequence is a contiguous stretch of chromosome DNA that spans all of the exons of the gene and includes transcribed and non transcribed stretches. For this ID use either the NCBI genomic nucleotide RefSeq IDs with their version number (see: NCBI.NLM.NIH.Gov/RefSeq) or use the LRG identifiers, without transcript (t or p) extensions -- when they become available. (See- Report sponsored by GEN2PHEN at the European Bioinformatics Institute at Hinxton UK April 24-25, 2008). The NCI RefSeq genomic IDs are distinguished by a prefix of"NG" for genes from the nuclear chromosomes and prefix of "NC" for genes from mitochondria. The LRG Identifiers have a prefix of "LRG_" Mitochondrial genes are not in the scope of LRG | 0..1 | display | |
| Struct var HGVS name | 0..1 | string | |
| The name of a structural variant reported using HGVS nomenclature. | 0..1 | display | |
| Ref nucleotide | 0..1 | string | |
| Reference values ("normal") examined within the Reference Sequence. This is used in a genotyping test to define the reference and variable nucleotide strings. That is if the sequence variation is an insertion, then Reference Nucleotide will be blank and Variable Nucleotide will contain the inserted nucleotides. In contrast, if the sequence variation is a deletion, then the Reference Nucleotide will contain the deleted nucliotieds, and the Variable Nucleotide will be blank. | 0..1 | display | |
| Gen allele loc ID | 0..1 | string | |
| The variant start-end location is the first genomic position in the reference allele that contains a change from the reference allele. For example, for the simple variant NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys), the variant exact start-end location is Chr3: 128906220 on Assembly GRCh38. [http://www.ncbi.nlm.nih.gov/clinvar/variation/30880/] | 0..1 | display | |
| Alt allele | 0..1 | string | |
| The genomic alternate allele is the contiguous segment of DNA in the test sample that differs from the reference allele at the same location and thus defines a variant. | 0..1 | display | |
| Haplotype name Bld/T | 0..1 | string | |
| dbSNP ID | 0..1 | string | |
| The unique identifier for the variant represented as a small nucleotide polymorphism (SNP). The dbSNP ID is used routinely as the base identifier in pharmacogenomics as well as arrCGH studies. For example, for the simple variant NM_014049.4(ACAD9):c.1249C>T (p.Arg417Cys), the dbSNP ID is 368949613. [http://www.ncbi.nlm.nih.gov/clinvar/variation/30880/] | 0..1 | display | |
| CIGAR var ID | 0..1 | string | |
| This term is used to report the unique ID from CIGAR, a syntax for describing variation that is use most frequently during alignment in sequencing studies. | 0..1 | display | |
| Cyto loc ID | 0..1 | string | |
| Genomic source class | 0..1 | choice | Options: 8 options |
| The genomic class of the specimen being analyzed: Germline for inherited genome, somatic for cancer genome, and prenatal for fetal genome. | 0..1 | display | |
| Struct var analysis method | 0..1 | choice | Options: 23 options |
| The method used for analyzing chromosome structural variation, such as FISH, arrCGH, sequencing, and MLPA. | 0..1 | display | |
| Gene dis seq var interp-Imp | 0..1 | choice | Options: 5 options |
| Single DNA marker or individual allele interpretation in the context of the assessed genetic disease. Prior to the LOINC release 2.56 (June 2016), the answer list was updated per the recommendations of the American College of Medical Genetics (ACMG). The previous answer list number was LL603-2, and two of the answer strings and LA codes are the same in the new list (pathogenic and benign). In the new answer list, the presumed pathogenic, unknown significance and presumed benign answers from LL603-2 have been replaced by likely pathogenic, uncertain significance and likely benign. The answer strings and their respective LA codes from LL603-2 remain valid. | 0..1 | display | |
| Genetic var assess | 0..1 | choice | Options: 4 options |
| Where testing scenarios are intended to assess the prescence or absence of a known set of DNA variants (e.g. tumor profiling using genotyping technology), then the Genetic Variant Assessment is used in conjunction with answer list supports structured communication of these findings. Of note, 'No Call' is different from 'Absent', because 'No Call' did not result in the determination of the marker's presents or absents. This may be due to test failure or specimen specific context which renders the test ineffective. | 0..1 | display | |
| Prob assoc phenotype | 0..1 | string | |
| The possible phenotype associated with the genetic variant found in this study. | 0..1 | display | |
| Allelic state | 0..1 | choice | Options: 5 options |
| The level of occurrence of a single DNA Marker within a set of chromosomes. Heterozygous indicates the DNA Marker is only present in one of the two genes contained in homologous chromosomes. Homozygous indicates the DNA Marker is present in both genes contained in homologous chromosomes. Hemizygous indicates the DNA Marker exists in the only single copy of a gene in a non-homologous chromosome (The male X and Y chromosome are non-homologous). Hemiplasmic indicates that the DNA Marker is present in some but not all of the copies of mitochondrial DNA. Homoplasmic indicates that the DNA Maker is present in all of the copies of mitochondrial DNA. | 0..1 | display | |
| Sample VAF | 0..1 | decimal | |
| The fraction of all reads in a study sample at a given genomic locus that identify the allele (variant) in question. For homozygotes it will be close to 1.0; for heterozygotes it will be close to 0.5. It can be less than 0.5 in the case of mosaics or multiple chromosome, or mixtures of tumor cells and normal cells. This measure is an attribute of the variant and applies when the method is a Next Generation Sequencing (NGS) or similar. Such methods provide many reads from the sample for each locus. To report population allelic frequency, see [LOINC: 92821-8]. Allelic frequency is usually reported as a decimal fraction for both Sample Variant Allelic Frequency and Population Allelic Frequency, although it is occasionally reported as a percent. Special care/caution should be taken when reporting and converting to a decimal fraction. | 0..1 | display | |
| Allelic read depth | 0..1 | decimal | |
| Allelic phase | 0..1 | choice | Options: 9 options |
| Basis allelic phase | 0..1 | choice | Options: 4 options |
| This panel is used to report the information associated with a simple genetic variant, such as a single nucleotide change. It should not be used to report information related to structural variants. | 0..1 | display | |
| Documentation for this format | ||||
Options Sets
Answer options for /81306-3/51968-6
Answer options for /81306-3/83006-7
Answer options for /81306-3/62374-4
Answer options for /81250-3/83005-9
Answer options for /81250-3/48019-4
Answer options for /81250-3/48006-1
Answer options for /81250-3/48002-0
Answer options for /81250-3/81304-8
Answer options for /81250-3/53037-8
Answer options for /81250-3/69548-6
Answer options for /81250-3/53034-5
Answer options for /81250-3/82120-7
Answer options for /81250-3/82309-6
Answer options for /81251-1/81263-6
Answer options for /81251-1/53037-8
Answer options for /81251-1/53034-5
Answer options for /81251-1/82309-6
Answer options for /81251-1/81250-3/83005-9
Answer options for /81251-1/81250-3/48019-4
Answer options for /81251-1/81250-3/48006-1
Answer options for /81251-1/81250-3/48002-0
Answer options for /81251-1/81250-3/81304-8
Answer options for /81251-1/81250-3/53037-8
Answer options for /81251-1/81250-3/69548-6
Answer options for /81251-1/81250-3/53034-5
Answer options for /81251-1/81250-3/82120-7
Answer options for /81251-1/81250-3/82309-6
Answer options for /82118-1/53040-2
Answer options for /82118-1/51961-1
Answer options for /82118-1/83009-1
Answer options for /82118-1/82117-3/82116-5
Answer options for /83011-7/81250-3/83005-9
Answer options for /83011-7/81250-3/48019-4
Answer options for /83011-7/81250-3/48006-1
Answer options for /83011-7/81250-3/48002-0
Answer options for /83011-7/81250-3/81304-8
Answer options for /83011-7/81250-3/53037-8
Answer options for /83011-7/81250-3/69548-6
Answer options for /83011-7/81250-3/53034-5
Answer options for /83011-7/81250-3/82120-7
Answer options for /83011-7/81250-3/82309-6
IG © 2024+ Manchester University NHS Foundation Trust. Package north-west.england.nhs.uk#0.0.1 based on FHIR 4.0.1. Generated 2025-04-20
Links: Table of Contents |
QA Report
| New Issue | Issues
Version History |
![]() |
Propose a change
|
Propose a change